LAL-D affects people of all ages with clinical manifestations from infancy through adulthood. 3 Infants with LAL-D can face rapid disease progression over a period of weeks that is typically fatal within a matter of months. The median age of death in these patients is 3.7 months. 4
LAL-D Symptoms
- Multi-organ damage
- Cardiovascular disease manifestations including dyslipidemia, accelerated atherosclerosis, coronary artery disease
- Liver damage including fibrosis, cirrhosis, and failure
- Failure to thrive and premature death
LAL-D* is a rare, inherited lysosomal storage disorder caused by pathogenic variants of the LIPA gene 1,2
In healthy individuals, LAL processes CEs and TGs as part of cholesterol homeostasis.3 In LAL-D, systemic lysosomal lipid accumulation leads to progressive multisystem organ damage. 1-3
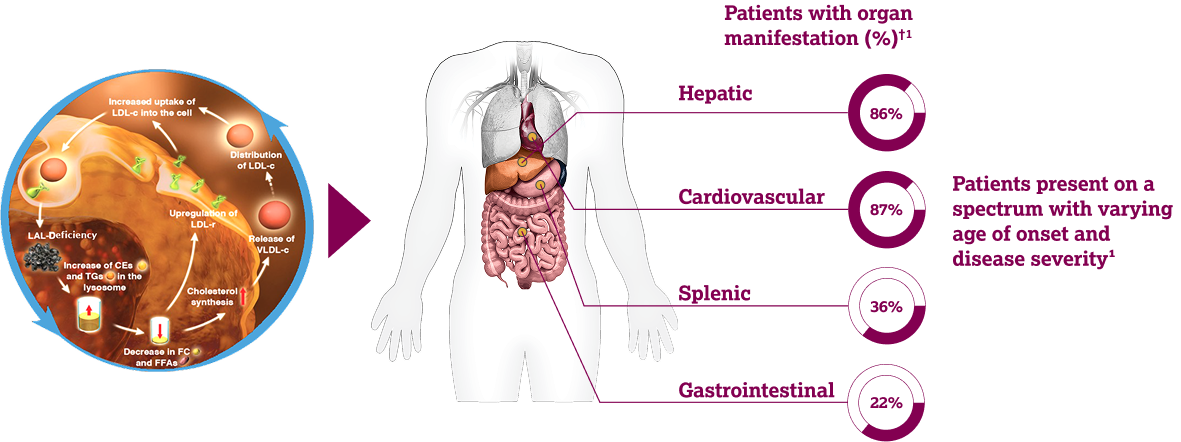
*LAL-D was previously known as Wolman disease (infants) and
Cholesteryl Ester Storage Disease (children and adults).
1,2
†Based on a literature review of 97 studies which evaluated 55
genotyped patients with LAL-D in a cohort of 135 patients with LAL-D
caused by deficient LAL activity. Age range: 0–68 years (median age
5 years).
1
CE, cholesteryl ester; FC, free cholesterol; FFA, free fatty acid;
LAL, lysosomal acid lipase; LAL-D, lysosomal acid lipase deficiency;
LDL-c, low-density lipoprotein cholesterol; LDL-r, low-density
lipoprotein receptor; LIPA, lipase A; TG, triglyceride; VLDL-c,
very-low-density lipoprotein cholesterol.
- Bernstein DL, et al. J Hepatol. 2013;58(6):1230–1243; 2; Reiner Ž, et al. Atherosclerosis. 2014;235(1):21–30; 3.Kohli R, et al. Mol Genet Metab. 2020;129(2):59–66.
- Jones, Simon A., et al. "Rapid progression and mortality of lysosomal acid lipase deficiency presenting in infants." Genetics in Medicine 18.5 (2016): 452-458.
- Hepatomegaly 1-6
- Hepatic injury 1,4
- ↑ ALT 1,3,4,6
- ↑ AST 1-3
- Microvesicular or mixed steatosis 1,3,4
- Fibrosis/micronodular cirrhosis 1-4,6
- Portal hypertension 1,2,4
- Liver failure 1,3-5
- Dyslipidaemia 1,2,4,6
- ↑ LDL-c 1,2,4,6
- ↓ HDL-c 1,2,4
- Accelerated atherosclerosis 1-4
- Coronary artery disease 1,3,4
- Stroke 1,3,4
- Myocardial infarction 1,4
- Splenomegaly 1-4,6,7
- Anaemia 4
- Thrombocytopenia 4
- Risk of traumatic rupture and/or splenectomy 4,7
- Intestinal lipid accumulation 4
- Abdominal pain 3,4
- Malabsorption 3-5
- Growth failure 3,4,8
ALT, alanine aminotransferase; AST, aspartate aminotransferase; HDL-c, high-density lipoprotein cholesterol; LDL-c, low-density lipoprotein cholesterol.
- Bernstein DL, et al. J Hepatol. 2013;58(6):1230–1243. 2. Kohli R, et al. Mol Genet Metab. 2020;129(2):59–66. 3. Reiner Ž, et al. Atherosclerosis. 2014;235(1):21–30. 4. Strebinger G, et al. Hepat Med. 2019;11:79–88.
- Pericleous M, et al. Lancet Gastroenterol Hepatol. 2017;2(9):670–679. 6. Burton BK, et al. J Pediatr Gastroenterol Nutr. 2015;61(6):619–625. 7. Oxford Medicine. Splenomegaly. Available at https://oxfordmedicine.com/view/10.1093/med/9780190862800.001.0001/med-9780190862800-chapter-31?rskey=2Z9YL6&result=1. Accessed October 2022. 8 Jones S, et al. Genet Med. 2016;18(3):452–458.
The signs and symptoms of LAL-D can overlap clinically and biochemically with other conditions 1-3
In healthy individuals, LAL processes CEs and TGs as part of cholesterol homeostasis. 3 In LAL-D, systemic lysosomal lipid accumulation leads to progressive multisystem organ damage. 1-3

*Not an exhaustive list.
†NAFLD is now known as metabolic dysfunction-associated steatotic
liver disease (MASLD).
‡Elevated LDL-c defined as ≥3.4 mmol/L (130 mg/dL) in children and
≥4.1 mmol/L (160 mg/dL) in adults.
ALT, alanine aminotransferase; FH, familial hypercholesterolemia;
FCH, familial combined hyperlipidemia; LAL-D, lysosomal acid lipase
deficiency; LDL-c, low-density lipoprotein cholesterol; NAFLD,
non-alcoholic fatty liver disease; NASH, non-alcohol related
steatohepatitis.
- Reiner Z, et al. Atherosclerosis. 2014;235:21–30; 2. Roberts EA, et al. Hepatology. 2008;47:2089–2111; 3. Kaur J. Cardiol Res Pract. 2014;943162; 4. Burton BK, et al. J Pediatr Gastroenterol Nutr. 2015;61(6):619–625; 5. Camarena C, et al. Med Clin (Barc). 2017;148(8):429.e1–429.e10.