In HPP, an early diagnosis can avoid inappropriate management that might worsen signs and symptoms, or even lead to complications of the disease1
- Hypophosphatasia (HPP) may have an evolving clinical burden that often remains high throughout childhood and adulthood1-3
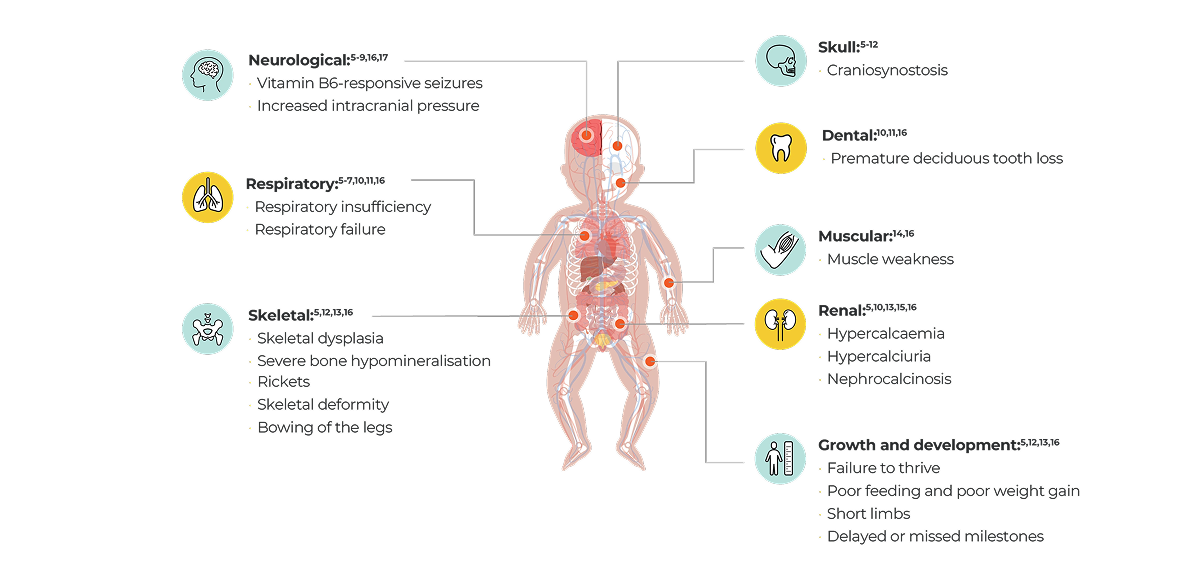
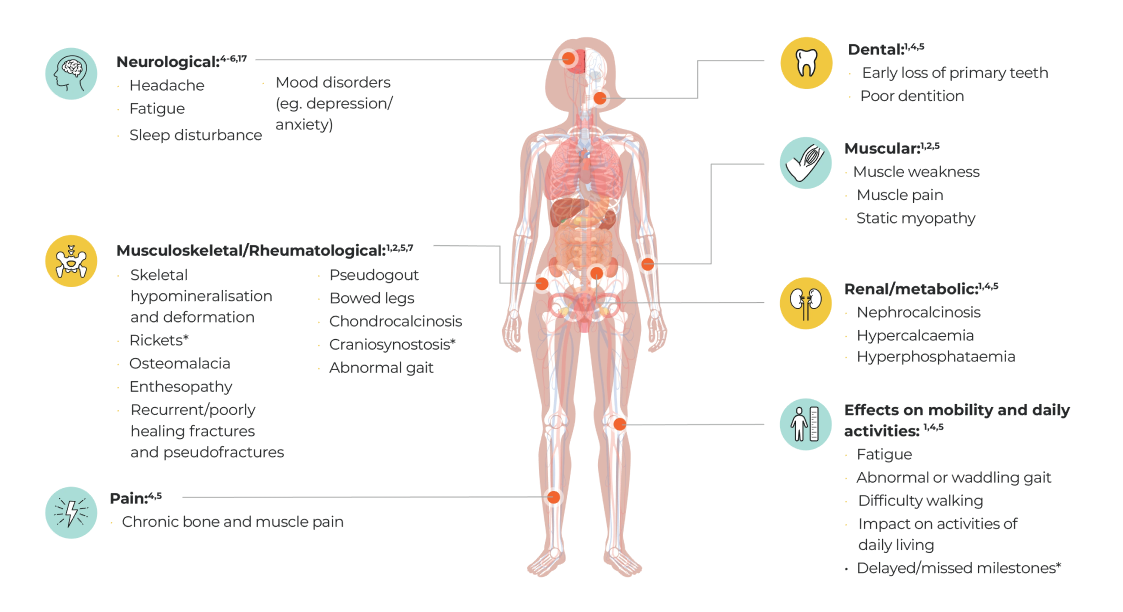
- HPP is often associated with a high clinical burden regardless of age at first manifestation.4 While HPP is frequently diagnosed during childhood, it may not be diagnosed until adulthood in some patients, even though patients may have had symptoms for years.1,2
- The signs and symptoms may change and accumulate over a patient’s lifetime with the disease manifesting differently in adults and children.4,5
- Children
- Adults
HPP is often misdiagnosed and confused with other bone or rheumatic
diseases
Do you know how HPP
can be diagnosed?
Learn more
- Högler W, et al. Diagnostic delay is common among patients with hypophosphatasia: initial findings from a longitudinal, prospective, global registry. BMC Musculoskeletal Discord. 2019;20(1):80.
- Conti F, et al. Hypophosphatasia: clinical manifestation and burden of disease in adult patients. Clin Cases Miner Bone Metab. 2017;14(2):230–234.
- Seefried L, et al. Burden of Illness in Adults With Hypophosphatasia: Data From the Global Hypophosphatasia Patient Registry. J Bone Miner Res. 2020;35(11):217 1–2178.
- Szabo S, et al. Frequency and age at occurrence of clinical manifestations of disease in patients with hypophosphatasia: a systematic literature review. Orphanet J Rare Dis. 2019;14(1):85.
- Khan et al. Hypophosphatasia diagnosis: current state of the art and proposed diagnostic criteria for children and adults. Osteoporosis International. 2024;35:431–438.
- Bishop N, et al. Transformative therapy in hypophosphatasia. Arch Dis Child. 2016;101:514–515.
- Whyte MP, et al. Natural History of Perinatal and Infantile Hypophosphatasia: A Retrospective Study. J Pediatr. 2019;209:116–124.e114;
- Surtees R, et al. Inborn errors affecting vitamin B6 metabolism. Future Neurol. 2006;1:615–620.
- Collmann H, et al. Neurosurgical aspects of childhood hypophosphatasia. Childs Nerv Syst. 2009;25:217–223.
- Bangura A, et al. Hypophosphatasia: Current Literature for Pathophysiology, Clinical Manifestations, Diagnosis, and Treatment. Cureus. 2020;12(6):e8594.
- Bloch-Zupan A. Hypophosphatasia: diagnosis and clinical signs - a dental surgeon perspective. Int J Paediatr Dent. 2016;26:426–438.
- Rockman-Greenberg C. Hypophosphatasia. Pediatr Endocrinol Rev. 2013;10(Suppl 2):380–388.
- Mohn A, et al. Hypophosphatasia in a child with widened anterior fontanelle: lessons learned from late diagnosis and incorrect treatment. Acta Paediatr. 2011;100:e43–e46.
- Rush ET, et al. Burden of disease in pediatric patients with hypophosphatasia: results from the HPP Impact Patient Survey and the HPP Outcomes Study Telephone interview. Orphanet J Rare Dis. 2019;14:201.
- Linglart A, Biosse-Duplan M. Hypophosphatasia. Curr Osteoporos Rep. 2016;14:95–105.
- Martos-Moreno, et al. Clinical Profiles of Children with Hypophosphatasia prior to Treatment with Enzyme Replacement Therapy: An Observational Analysis from the Global HPP Registry. Horm Res Paediatr. 2024;97(3):233–242.
- Colazo JM. Neurological symptoms in Hypophosphatasia. Osteoporos Int. 2019;30(2):469–480.